Genomic Feature Analysis
Pipeline overview:
- Make master genome feature bed file
- download feature files (.gff and beds) from OSF
- concatenate files
- Use bedtools intersect to match DMRs to features in master genome feature file
- Generate background regions
- find all CpGs covered by at least 3/4 samples per treatment group
- then find CpGs common to all groups within each comparison (all ambient samples, all day 10 samples, etc.)
- bin features in master genome feature bed file (2 kb)
- the purpose of this is to make background regions more similar to DMRs; DMRs are between 6 and ~2kb. Some features are much larger (e.g. intergenic region can be > 10kb).
- determine binned features that overlap with covered CpGs
- filter features for those with at least 3 covered CpGs
- determine if the number of DMRs within certain feature categories is significantly different than the number of covered regions within certain feature categories
- read data into R
- create contingency tables for each comparison
Scripts
- 20200403_feature_analysis.ipynb: Jupyter notebook that performs pipeline steps 1-3
- 20200406_feat_stats.Rmd: R markdown script that performs Chi square statistical analysis and visualization of genomic feature overlap
Script output
Data
- Bed file from concatenated gffs: https://gannet.fish.washington.edu/metacarcinus/Pgenerosa/analyses/20200403/Panopea-generosa-v1.0.a4.uniq.bed
- Binned bed file from concatenated gffs: https://gannet.fish.washington.edu/metacarcinus/Pgenerosa/analyses/20200406/Panopea-generosa-v1.0.a4.2Kbin.bed
- Bed files with background regions for each comparison
- https://gannet.fish.washington.edu/metacarcinus/Pgenerosa/analyses/20200406/amb.3xCpG.allgrps.bed
- https://gannet.fish.washington.edu/metacarcinus/Pgenerosa/analyses/20200406/day10.3xCpG.allgrps.bed
- https://gannet.fish.washington.edu/metacarcinus/Pgenerosa/analyses/20200406/day135.3xCpG.allgrps.bed
- https://gannet.fish.washington.edu/metacarcinus/Pgenerosa/analyses/20200406/day145.3xCpG.allgrps.bed
- DMRs overlapping with features:
- Background regions overlapping with features:
- https://gannet.fish.washington.edu/metacarcinus/Pgenerosa/analyses/20200406/amb_features.3CpG.txt
- https://gannet.fish.washington.edu/metacarcinus/Pgenerosa/analyses/20200406/day10_features.3CpG.txt
- https://gannet.fish.washington.edu/metacarcinus/Pgenerosa/analyses/20200406/day135_features.3CpG.txt
- https://gannet.fish.washington.edu/metacarcinus/Pgenerosa/analyses/20200406/day145_features.3CpG.txt
- other intermediate files can be found here: https://gannet.fish.washington.edu/metacarcinus/Pgenerosa/analyses/20200406/ and https://github.com/shellywanamaker/Shelly_Pgenerosa/tree/master/analyses/20200406_anno
Plots
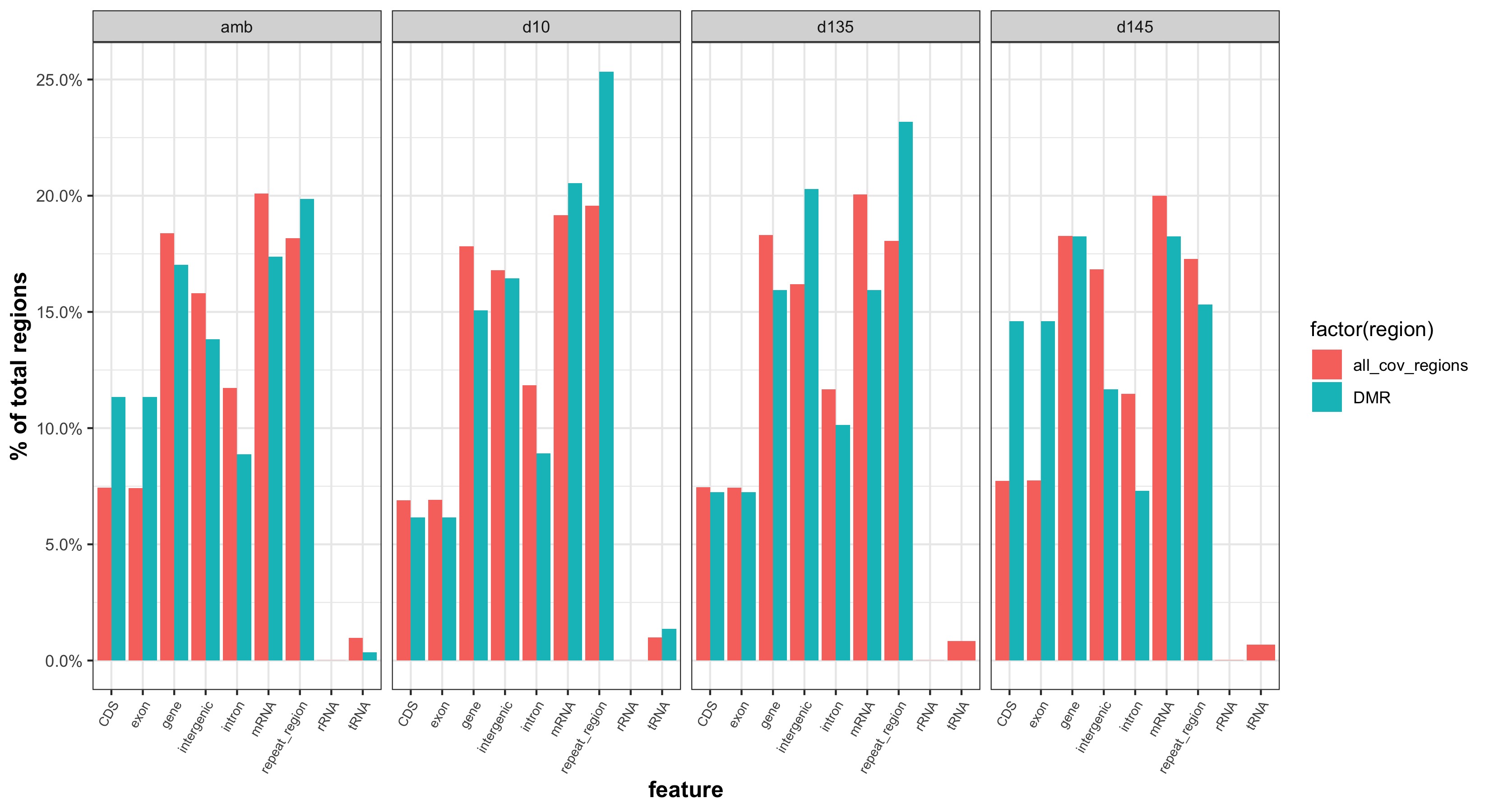
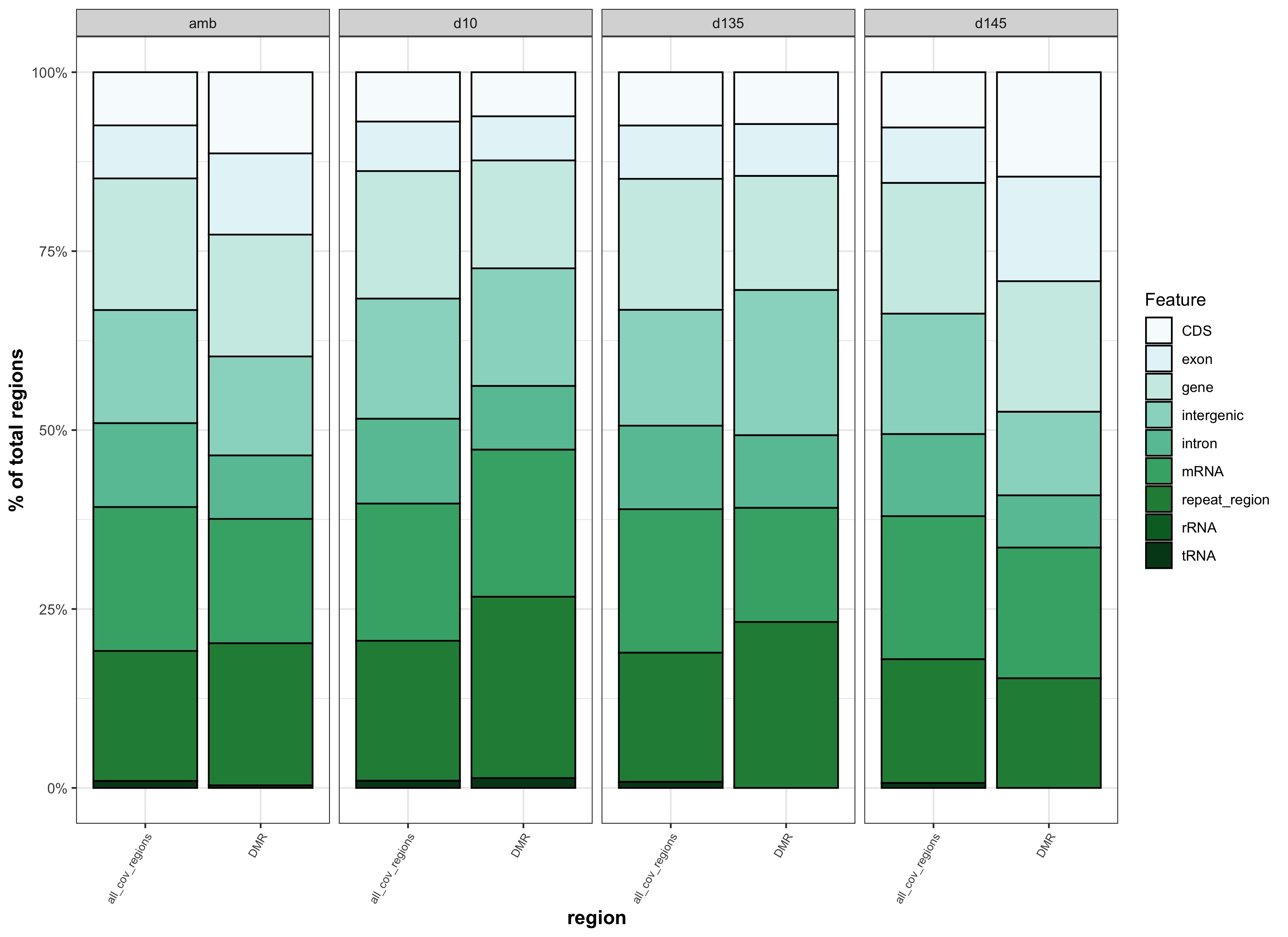
Tables
Chi sq table generated by script
Next steps
- reduce the number of features
- remove rRNA since no data overlap with these
- consolidate exon, CDS, gene, mRNA, etc.
- add new features
- UTR
- putative promoter
- re-run analyses with updated features