Purpose
Examine relationships between methylation and expression
Methods
Jupyter nb here: combined_methylation-analysis.ipynb
Combined Exon and Gene Methylation Analysis
This document presents a comprehensive analysis of exon and gene methylation, including:
- Histograms and heatmaps of exon positions by methylation level
- Exon and gene methylation density by individual and season
- Correlation plots of gene expression and methylation
- Coefficient of variation (CV) of expression vs. mean methylation (gene and exon)
All code and plots are included for reproducibility and blog post preparation.
Import Required Libraries
We will use pandas, numpy, matplotlib, seaborn, and re for data analysis and visualization.
Combined Exon and Gene Methylation Analysis
This document presents a comprehensive analysis of exon and gene methylation, including:
- Histograms and heatmaps of exon positions by methylation level
- Exon and gene methylation density by individual and season
- Correlation plots of gene expression and methylation
- Coefficient of variation (CV) of expression vs. mean methylation (gene and exon)
All code and plots are included for reproducibility and blog post preparation.
Import Required Libraries
We will use pandas, numpy, matplotlib, seaborn, and re for data analysis and visualization.
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import seaborn as sns
import re
{bash}
#create bed files from gffs
awk -F "\t" '{if($3!="")print $1"\t"$4"\t"$5"\t"$9}' Porites_evermanni_validated.gff3 > Porites_evermanni_validated.gff.bed
awk -F "\t" '{if($3!="")print $1"\t"$4"\t"$5"\t"$9}' Pocillopora_meandrina_HIv1.genes-validated.gff > Pocillopora_meandrina_HIv1.gff.bed
awk -F "\t" '{if($3!="")print $1"\t"$4"\t"$5"\t"$3";"$9}' Apulcra-genome.gff > Apulcra-genome.gff.bed
#create 10x perc methylation files for each sample for each species. This code matches CpGs to features in the gff bed file and only keeps loci that have >= 10 counts. And then it sums all unmethylated loci within a feature, and sums all methylated loci within a feature, and calculates % methylation from that.
#for Peve
for f in *.CpG_report.merged_CpG_evidence.cov.gz
do
base=$(basename "$f" .CpG_report.merged_CpG_evidence.cov.gz)
echo "Processing $base..."
intersectBed -wa -wb -a /gscratch/srlab/strigg/GENOMES/Porites_evermanni_validated.gff.bed -b $f | awk '{if($9+$10>=10)print $0}' | awk '{g = $4;meth[g] += $9;unmeth[g] += $10}END{for(g in meth){total = meth[g] + unmeth[g];pct = (meth[g] / total * 100);print g"\t"meth[g]"\t"unmeth[g]"\t"pct}}' > "${base}_perc_meth.txt"
done
#for Ptua
for f in *.CpG_report.merged_CpG_evidence.cov.gz
do
base=$(basename "$f" .CpG_report.merged_CpG_evidence.cov.gz)
echo "Processing $base..."
intersectBed -wa -wb -a /gscratch/srlab/strigg/GENOMES/Pocillopora_meandrina_HIv1.gff.bed -b $f | awk '{if($9+$10>=10)print $0}' | awk '{g = $4;meth[g] += $9;unmeth[g] += $10}END{for(g in meth){total = meth[g] + unmeth[g];pct = (meth[g] / total * 100);print g"\t"meth[g]"\t"unmeth[g]"\t"pct}}' > "${base}_perc_meth.txt"
done
#for Apulcra
for f in *.CpG_report.merged_CpG_evidence.cov.gz
do
base=$(basename "$f" .CpG_report.merged_CpG_evidence.cov.gz)
echo "Processing $base..."
intersectBed -wa -wb -a /gscratch/srlab/strigg/GENOMES/Apulcra-genome.gff.bed -b $f | awk '{if($9+$10>=10)print $0}' | awk '{g = $4;meth[g] += $9;unmeth[g] += $10}END{for(g in meth){total = meth[g] + unmeth[g];pct = (meth[g] / total * 100);print g"\t"meth[g]"\t"unmeth[g]"\t"pct}}' > "${base}_perc_meth.txt"
done
# Merge all perc_meth.txt files, filtering for ID=gene
echo "Merging files..."
for f in *_perc_meth.txt
do
base=$(basename "$f" _perc_meth.txt)
grep "gene;" "$f" | awk -v sample="$base" '{print sample"\t"$1"\t"$4}'
done | awk '{
sample=$1;
gene=$2;
val=$3;
if(!seen[sample]) {
samples[++n]=sample;
seen[sample]=1;
}
data[gene][sample]=val;
genes[gene]=1;
}
END {
printf "gene";
for(i=1; i<=n; i++) printf "\t%s", samples[i];
printf "\n";
for(gene in genes) {
printf "%s", gene;
for(i=1; i<=n; i++) {
printf "\t%s", (data[gene][samples[i]] ? data[gene][samples[i]] : "NA");
}
printf "\n";
}
}' > Apul_merged_perc_meth.txt
echo "Done! Output in Apul_merged_perc_meth.txt"
#### Exons
echo "Merging files..."
for f in *_perc_meth.txt
do
base=$(basename "$f" _perc_meth.txt)
grep "exon;" "$f" | awk -v sample="$base" '{print sample"\t"$1"\t"$4}'
done | awk '{
sample=$1;
gene=$2;
val=$3;
if(!seen[sample]) {
samples[++n]=sample;
seen[sample]=1;
}
data[gene][sample]=val;
genes[gene]=1;
}
END {
printf "gene";
for(i=1; i<=n; i++) printf "\t%s", samples[i];
printf "\n";
for(gene in genes) {
printf "%s", gene;
for(i=1; i<=n; i++) {
printf "\t%s", (data[gene][samples[i]] ? data[gene][samples[i]] : "NA");
}
printf "\n";
}
}' > Apul_exon_merged_perc_meth.txt
echo "Done! Output in Apul_exon_merged_perc_meth.txt"
### Peve #####
echo "Merging files..."
for f in *_perc_meth.txt
do
base=$(basename "$f" _perc_meth.txt)
grep "ID=gene" "$f" | awk -v sample="$base" '{print sample"\t"$1"\t"$4}'
done | awk '{
sample=$1;
gene=$2;
val=$3;
if(!seen[sample]) {
samples[++n]=sample;
seen[sample]=1;
}
data[gene][sample]=val;
genes[gene]=1;
}
END {
printf "gene";
for(i=1; i<=n; i++) printf "\t%s", samples[i];
printf "\n";
for(gene in genes) {
printf "%s", gene;
for(i=1; i<=n; i++) {
printf "\t%s", (data[gene][samples[i]] ? data[gene][samples[i]] : "NA");
}
printf "\n";
}
}' > Peve_merged_perc_meth.txt
echo "Done! Output in Peve_merged_perc_meth.txt"
--------------------
### Exons ###
echo "Merging files..."
for f in *_perc_meth.txt
do
base=$(basename "$f" _perc_meth.txt)
grep "ID=exon" "$f" | awk -v sample="$base" '{print sample"\t"$1"\t"$4}'
done | awk '{
sample=$1;
gene=$2;
val=$3;
if(!seen[sample]) {
samples[++n]=sample;
seen[sample]=1;
}
data[gene][sample]=val;
genes[gene]=1;
}
END {
printf "gene";
for(i=1; i<=n; i++) printf "\t%s", samples[i];
printf "\n";
for(gene in genes) {
printf "%s", gene;
for(i=1; i<=n; i++) {
printf "\t%s", (data[gene][samples[i]] ? data[gene][samples[i]] : "NA");
}
printf "\n";
}
}' > Peve_exon_merged_perc_meth.txt
echo "Done!"
---------------------
echo "Merging files..."
for f in *_perc_meth.txt
do
base=$(basename "$f" _perc_meth.txt)
grep "ID=gene" "$f" | awk -v sample="$base" '{print sample"\t"$1"\t"$4}'
done | awk '{
sample=$1;
gene=$2;
val=$3;
if(!seen[sample]) {
samples[++n]=sample;
seen[sample]=1;
}
data[gene][sample]=val;
genes[gene]=1;
}
END {
printf "gene";
for(i=1; i<=n; i++) printf "\t%s", samples[i];
printf "\n";
for(gene in genes) {
printf "%s", gene;
for(i=1; i<=n; i++) {
printf "\t%s", (data[gene][samples[i]] ? data[gene][samples[i]] : "NA");
}
printf "\n";
}
}' > Ptua_merged_perc_meth.txt
echo "Done! Output in Ptua_merged_perc_meth.txt"
----------------------
for f in *_perc_meth.txt
do
base=$(basename "$f" _perc_meth.txt)
grep "ID=exon" "$f" | awk -v sample="$base" '{print sample"\t"$1"\t"$4}'
done | awk '{
sample=$1;
gene=$2;
val=$3;
if(!seen[sample]) {
samples[++n]=sample;
seen[sample]=1;
}
data[gene][sample]=val;
genes[gene]=1;
}
END {
printf "gene";
for(i=1; i<=n; i++) printf "\t%s", samples[i];
printf "\n";
for(gene in genes) {
printf "%s", gene;
for(i=1; i<=n; i++) {
printf "\t%s", (data[gene][samples[i]] ? data[gene][samples[i]] : "NA");
}
printf "\n";
}
}' > Ptua_exon_merged_perc_meth.txt
-------------------
75% of samples (~1/2 genes)
#Peve
awk '{na_count = 0;for (i = 1; i <= NF; i++) {if ($i == "NA") {na_count++;}}if (na_count <= 9) {print;}}' Peve_merged_perc_meth.txt > Peve_merged_perc_meth_filt75.txt
awk '{na_count = 0;for (i = 1; i <= NF; i++) {if ($i == "NA") {na_count++;}}if (na_count <= 9) {print;}}' Peve_exon_merged_perc_meth.txt > Peve_exon_merged_perc_meth_filt75.txt
# Ptua
awk '{na_count = 0;for (i = 1; i <= 33; i++) {if ($i == "NA") {na_count++;}}if (na_count <= 8) {print;}}' your_file.txt
awk '{na_count = 0;for (i = 1; i <= 33; i++) {if ($i == "NA") {na_count++;}}if (na_count <= 8) {print;}}' Ptua_exon_merged_perc_meth.txt > Ptua_exon_merged_perc_meth_filt75.txt
#Apul
awk '{na_count = 0;for (i = 1; i <= NF; i++) {if ($i == "NA") {na_count++;}}if (na_count <= 10) {print;}}' Apul_merged_perc_meth.txt > Apul_merged_perc_meth_filt75.txt
awk '{na_count = 0;for (i = 1; i <= NF; i++) {if ($i == "NA") {na_count++;}}if (na_count <= 10) {print;}}' Apul_exon_merged_perc_meth.txt > Apul_exon_merged_perc_meth_filt75.txt
10K genes recip blast on mosaic: plot exp x meth:
create meth plots for genes
calcification ortho group expression matrix: these are from jill
## Load and Prepare Data
#We will load the annotated exon methylation data and extract exon positions for downstream analysis.
# Load annotated methylation table
exon_df = pd.read_csv('annotated_exon_methylation.csv', index_col=0)
def get_exon_pos(exon_name):
if pd.isnull(exon_name):
return None
m = re.search(r'-(\\d+)$', exon_name)
return int(m.group(1)) if m else None
if 'exon_position' not in exon_df.columns:
exon_df['exon_position'] = exon_df['exon_name'].apply(get_exon_pos)
exon_df = exon_df.dropna(subset=['exon_position'])
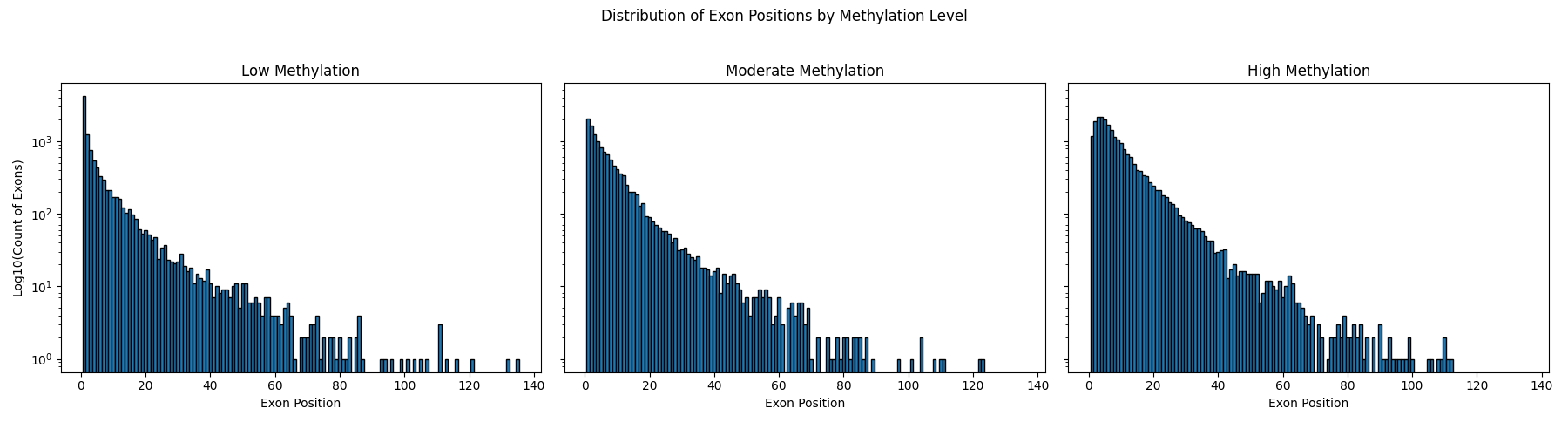
Histogram of Exon Positions by Methylation Level (Log Scale)
Visualize the distribution of exon positions for each methylation level using log-transformed counts.
import pandas as pd
import matplotlib.pyplot as plt
import re
# Load annotated methylation table
df = pd.read_csv('annotated_exon_methylation.csv', index_col=0)
# Extract exon position from exon_name (number after last dash)
def get_exon_pos(exon_name):
if pd.isnull(exon_name):
return None
m = re.search(r'-(\d+)$', exon_name)
return int(m.group(1)) if m else None
df['exon_position'] = df['exon_name'].apply(get_exon_pos)
# Drop missing positions
df = df.dropna(subset=['exon_position'])
# Plot histogram for each methylation level
levels = ['low', 'moderate', 'high']
fig, axes = plt.subplots(1, 3, figsize=(18, 5), sharey=True)
for ax, level in zip(axes, levels):
sub = df[df['methylation_level'] == level]
ax.hist(sub['exon_position'], bins=range(1, int(df['exon_position'].max())+2), align='left', edgecolor='black', log=True)
ax.set_title(f'{level.capitalize()} Methylation')
ax.set_xlabel('Exon Position')
if ax is axes[0]:
ax.set_ylabel('Log10(Count of Exons)')
plt.suptitle('Distribution of Exon Positions by Methylation Level')
plt.tight_layout(rect=[0, 0.03, 1, 0.95])
plt.savefig('exon_position_hist_by_methylation_level.png')
plt.show()
import pandas as pd
import seaborn as sns
import matplotlib.pyplot as plt
import numpy as np
import re
# Load your data
# Adjust the filename if needed
file = 'annotated_exon_methylation.csv'
df = pd.read_csv(file, index_col=0)
def get_exon_pos(exon_name):
if pd.isnull(exon_name):
return None
m = re.search(r'-(\d+)$', exon_name)
return int(m.group(1)) if m else None
if 'exon_position' not in df.columns:
df['exon_position'] = df['exon_name'].apply(get_exon_pos)
df = df.dropna(subset=['exon_position'])
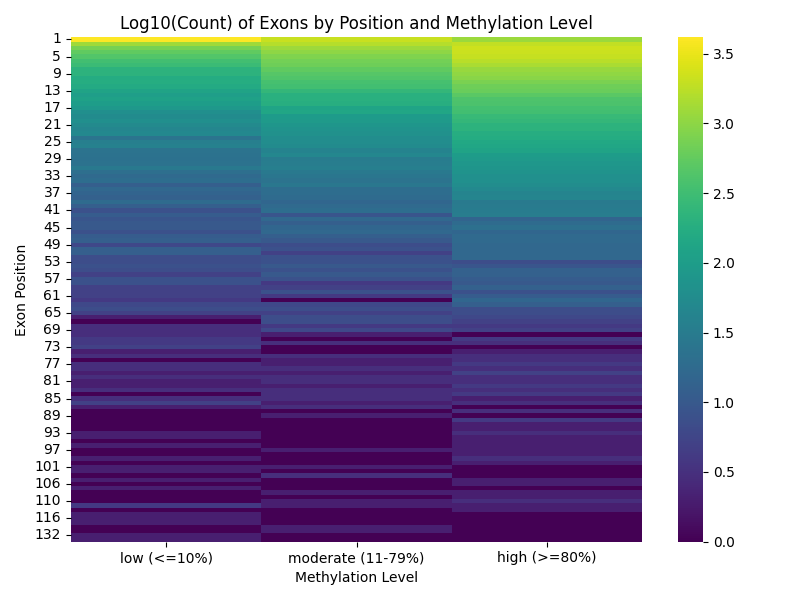
# Create a pivot table: rows=exon_position, columns=methylation_level, values=counts
level_order = ['low', 'moderate', 'high']
df['methylation_level'] = pd.Categorical(df['methylation_level'], categories=level_order, ordered=True)
heatmap_data = df.groupby(['exon_position', 'methylation_level']).size().unstack(fill_value=0)[level_order]
# Log-transform the counts for better visualization
heatmap_data_log = np.log10(heatmap_data + 1)
# Plot heatmap without annotation numbers
plt.figure(figsize=(8, 6))
ax = sns.heatmap(heatmap_data_log, cmap='viridis', annot=False)
plt.title('Log10(Count) of Exons by Position and Methylation Level')
# Custom x-axis labels
ax.set_xlabel('Methylation Level')
ax.set_ylabel('Exon Position')
ax.set_xticklabels([
'low (<=10%)',
'moderate (11-79%)',
'high (>=80%)'
])
plt.tight_layout()
plt.savefig('exon_position_heatmap_by_methylation_level.png')
plt.show()
#!/usr/bin/env python3
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import re
# Load gene expression data
expr_df = pd.read_csv('ptua-gene_count_matrix.csv', index_col=0)
# Load methylation data, fix gene name column
meth_df = pd.read_csv('Ptua_merged_perc_meth_filt75.txt', sep='\t')
meth_df.rename(columns={'gene': 'gene_id'}, inplace=True)
meth_df['gene_id'] = meth_df['gene_id'].str.replace('ID=', '', regex=False)
meth_df.set_index('gene_id', inplace=True)
# Find common genes
common_genes = expr_df.index.intersection(meth_df.index)
expr_common = expr_df.loc[common_genes]
meth_common = meth_df.loc[common_genes]
# Parse sample columns to get individual and timepoint
def parse_sample(sample):
m = re.match(r'(POC-\d+)-(TP\d)', sample)
if m:
return m.group(1), m.group(2)
return None, None
samples = [col for col in expr_common.columns if parse_sample(col)[0] is not None]
individuals = sorted(set(parse_sample(s)[0] for s in samples))
timepoints = sorted(set(parse_sample(s)[1] for s in samples))
colors = dict(zip(timepoints, ['#1f77b4', '#ff7f0e', '#2ca02c', '#d62728']))
# Load biomin genes
# Load biomin genes and add 'gene-' prefix for matching
biomin_df = pd.read_csv('ptua_biomin_counts.csv')
biomin_genes = set('gene-' + gid for gid in biomin_df['gene_id'])
individual_colors = dict(zip(individuals, plt.cm.tab20.colors[:len(individuals)]))
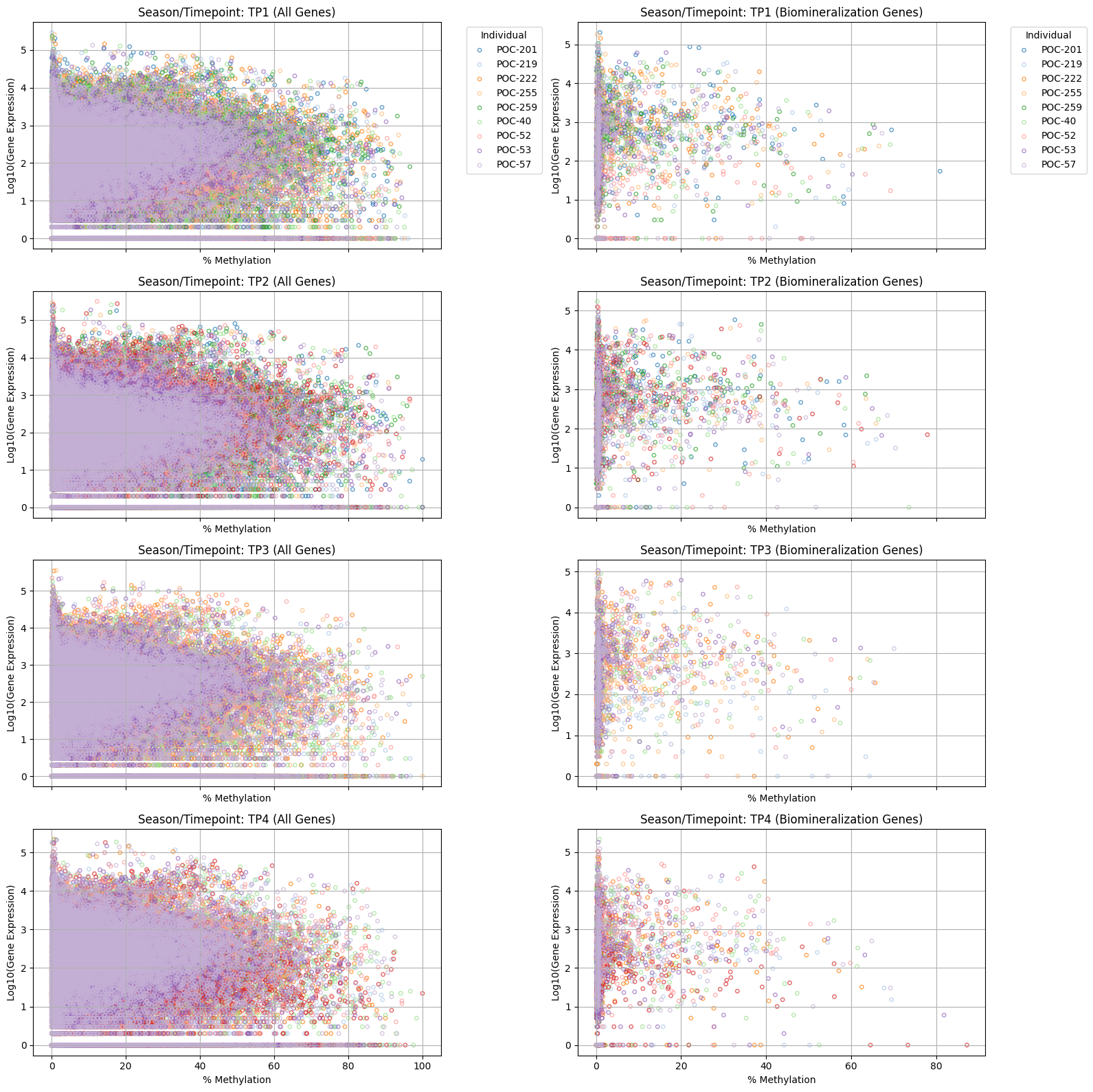
# 4 rows (seasons), 2 columns (all genes, biomin genes)
fig, axes = plt.subplots(len(timepoints), 2, figsize=(16, 4*len(timepoints)), sharex='col')
for i, tp in enumerate(timepoints):
# All genes subplot (col 0)
ax_all = axes[i, 0]
for ind in individuals:
sample_name = f"{ind}-{tp}"
if sample_name in expr_common.columns and sample_name in meth_common.columns:
expr_vals = expr_common[sample_name]
meth_vals = meth_common[sample_name]
log_expr = np.log10(expr_vals + 1)
ax_all.scatter(meth_vals, log_expr, alpha=0.7, label=ind, facecolors='none', edgecolors=individual_colors[ind], s=15)
ax_all.set_title(f"Season/Timepoint: {tp} (All Genes)")
ax_all.set_xlabel('% Methylation')
ax_all.set_ylabel('Log10(Gene Expression)')
ax_all.grid(True)
if i == 0:
ax_all.legend(title='Individual', bbox_to_anchor=(1.05, 1), loc='upper left')
# Biomin genes subplot (col 1)
ax_biomin = axes[i, 1]
for ind in individuals:
sample_name = f"{ind}-{tp}"
if sample_name in expr_common.columns and sample_name in meth_common.columns:
# Only keep biomin genes
expr_vals_bio = expr_common.loc[expr_common.index.isin(biomin_genes), sample_name]
meth_vals_bio = meth_common.loc[meth_common.index.isin(biomin_genes), sample_name]
log_expr_bio = np.log10(expr_vals_bio + 1)
ax_biomin.scatter(meth_vals_bio, log_expr_bio, alpha=0.7, label=ind, facecolors='none', edgecolors=individual_colors[ind], s=15)
ax_biomin.set_title(f"Season/Timepoint: {tp} (Biomineralization Genes)")
ax_biomin.set_xlabel('% Methylation')
ax_biomin.set_ylabel('Log10(Gene Expression)')
ax_biomin.grid(True)
if i == 0:
ax_biomin.legend(title='Individual', bbox_to_anchor=(1.05, 1), loc='upper left')
plt.tight_layout()
plt.savefig('correlation_plot_by_season_and_biomin.png', bbox_inches='tight')
plt.show()
#!/usr/bin/env python3
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
# Load gene expression data
expr_df = pd.read_csv('ptua-gene_count_matrix.csv', index_col=0)
# Load methylation data
meth_df = pd.read_csv('Ptua_merged_perc_meth_filt75.txt', sep='\t')
meth_df.rename(columns={'gene': 'gene_id'}, inplace=True)
meth_df['gene_id'] = meth_df['gene_id'].str.replace('ID=', '', regex=False)
meth_df.set_index('gene_id', inplace=True)
# Find common genes
common_genes = expr_df.index.intersection(meth_df.index)
expr_common = expr_df.loc[common_genes]
meth_common = meth_df.loc[common_genes]
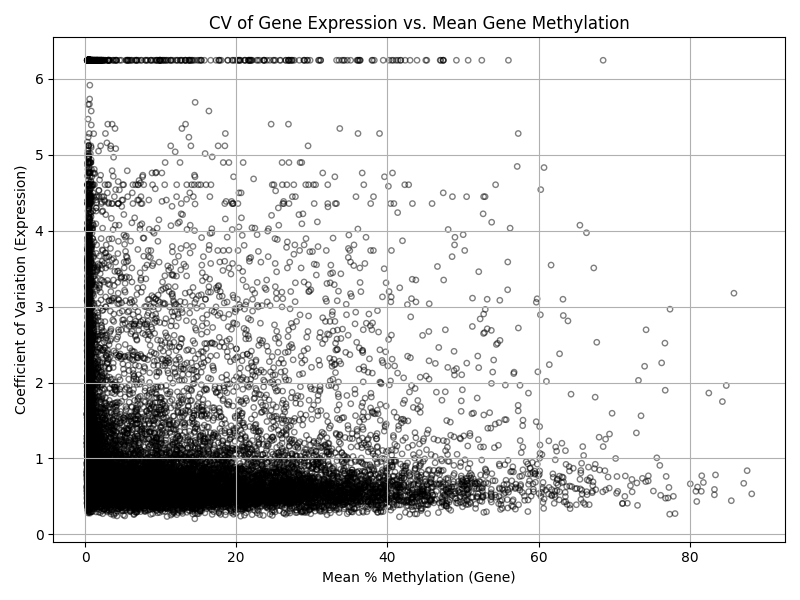
# Coefficient of variation (CV) for gene expression across samples
cv_expr = expr_common.std(axis=1) / expr_common.mean(axis=1)
# Mean methylation across samples
mean_meth = meth_common.mean(axis=1)
# Plot CV of expression vs. mean methylation
plt.figure(figsize=(8,6))
plt.scatter(mean_meth, cv_expr, alpha=0.5, edgecolors='k', facecolors='none', s=15)
plt.xlabel('Mean % Methylation (Gene)')
plt.ylabel('Coefficient of Variation (Expression)')
plt.title('CV of Gene Expression vs. Mean Gene Methylation')
plt.grid(True)
plt.tight_layout()
plt.savefig('cv_expression_vs_mean_methylation.png')
plt.show()
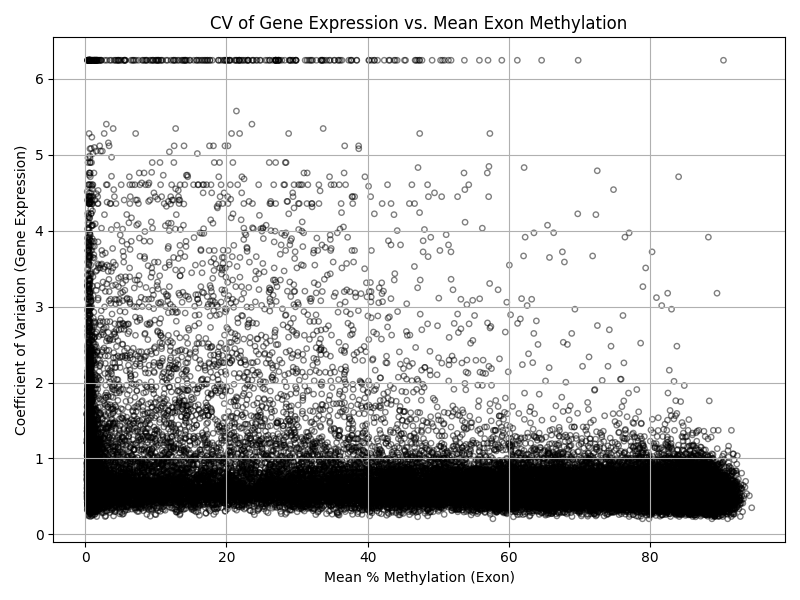
#!/usr/bin/env python3
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import re
# Load gene expression data
expr_df = pd.read_csv('ptua-gene_count_matrix.csv', index_col=0)
# Load exon methylation data
exon_meth_df = pd.read_csv('Ptua_exon_merged_perc_meth_filt75.txt', sep='\t')
# Parse exon names to extract parent gene
exon_meth_df['parent_gene'] = exon_meth_df['gene'].apply(lambda x: 'gene-' + re.search(r'Parent=mrna-([^;]+)', x).group(1) if re.search(r'Parent=mrna-([^;]+)', x) else None)
exon_meth_df = exon_meth_df.dropna(subset=['parent_gene'])
# Only keep exons whose parent gene is in expression data
genes_in_expr = set(expr_df.index)
exon_meth_df = exon_meth_df[exon_meth_df['parent_gene'].isin(genes_in_expr)]
# Get sample columns
sample_cols = [col for col in exon_meth_df.columns if re.match(r'POC-\d+-TP\d', col)]
# Mean % methylation for each exon
mean_exon_meth = exon_meth_df[sample_cols].mean(axis=1)
# CV of gene expression for parent gene
parent_gene_means = expr_df.loc[exon_meth_df['parent_gene']].mean(axis=1)
parent_gene_stds = expr_df.loc[exon_meth_df['parent_gene']].std(axis=1)
cv_expr = parent_gene_stds / parent_gene_means
# Combine into a DataFrame for filtering
# Reset index to avoid duplicate labels
plot_df = pd.DataFrame({
'mean_exon_meth': mean_exon_meth.values,
'cv_expr': cv_expr.values,
'parent_gene_means': parent_gene_means.values
})
# Filter: remove exons with zero or missing mean expression or mean methylation
plot_df = plot_df[(plot_df['mean_exon_meth'] > 0) & (plot_df['parent_gene_means'] > 0) & (~plot_df['mean_exon_meth'].isna()) & (~plot_df['cv_expr'].isna())]
# Plot: x = mean % methylation (exon), y = CV of gene expression
plt.figure(figsize=(8,6))
plt.scatter(plot_df['mean_exon_meth'], plot_df['cv_expr'], alpha=0.5, edgecolors='k', facecolors='none', s=15)
plt.xlabel('Mean % Methylation (Exon)')
plt.ylabel('Coefficient of Variation (Gene Expression)')
plt.title('CV of Gene Expression vs. Mean Exon Methylation')
plt.grid(True)
plt.tight_layout()
plt.savefig('cv_expression_vs_mean_exon_methylation.png')
plt.show()
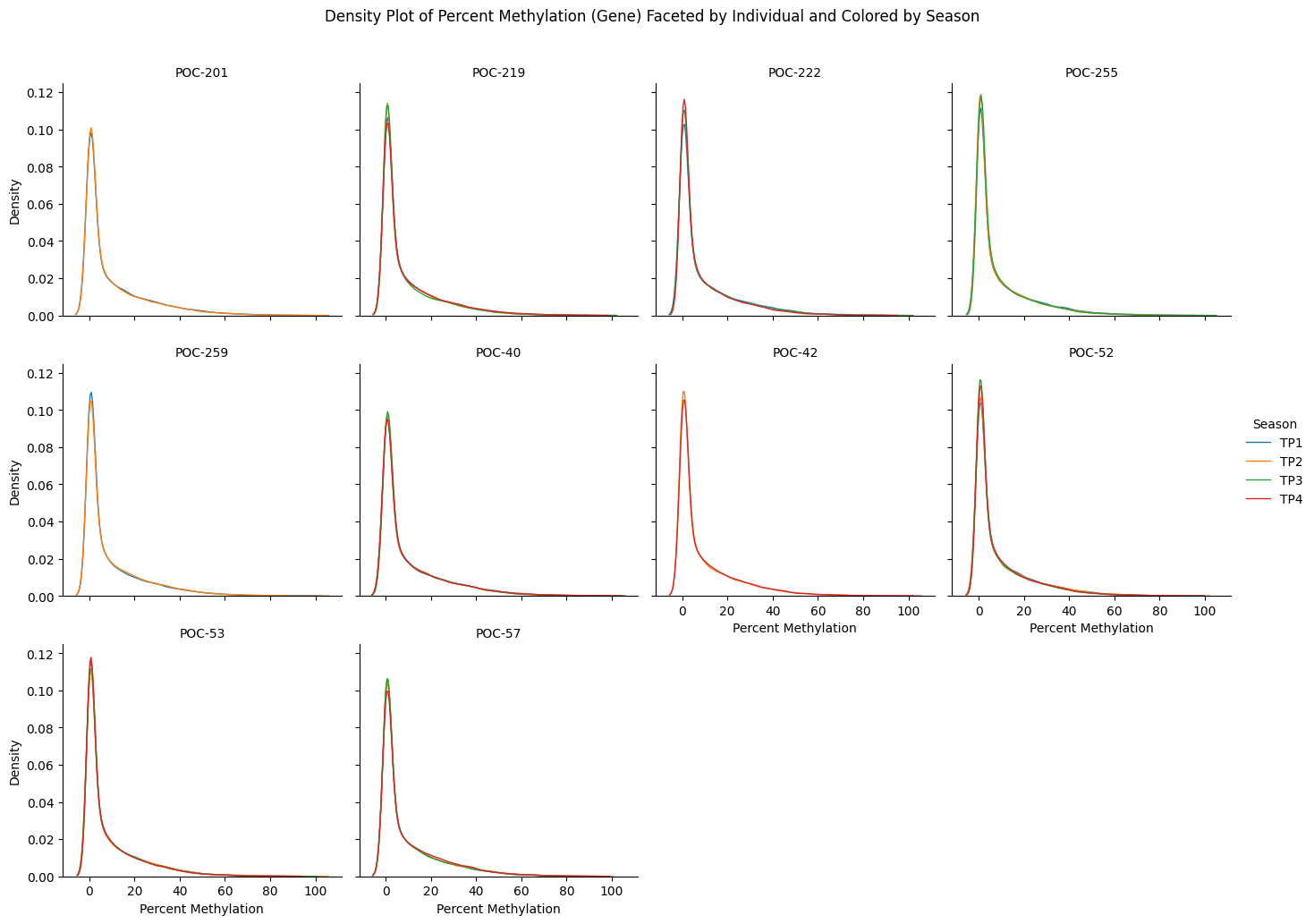
#!/usr/bin/env python3
import pandas as pd
import matplotlib.pyplot as plt
import seaborn as sns
import re
# Load methylation data
gene_meth_df = pd.read_csv('Ptua_merged_perc_meth_filt75.txt', sep='\t')
gene_meth_long = gene_meth_df.melt(id_vars=['gene'], var_name='Sample', value_name='Percent_Methylation')
gene_meth_long = gene_meth_long.dropna(subset=['Percent_Methylation'])
gene_meth_long['Individual'] = gene_meth_long['Sample'].apply(lambda x: re.sub(r'-TP\d+$', '', x))
gene_meth_long['Season'] = gene_meth_long['Sample'].apply(lambda x: re.search(r'(TP\d+)$', x).group(1) if re.search(r'(TP\d+)$', x) else 'NA')
g = sns.FacetGrid(gene_meth_long, col='Individual', col_wrap=4, sharex=True, sharey=True, height=3.5, hue='Season', palette='tab10')
g.map(sns.kdeplot, 'Percent_Methylation', fill=False, linewidth=1)
g.add_legend(title='Season')
g.set_titles(col_template="{col_name}")
g.set_axis_labels('Percent Methylation', 'Density')
plt.subplots_adjust(top=0.9)
g.fig.suptitle('Density Plot of Percent Methylation (Gene) Faceted by Individual and Colored by Season')
plt.savefig('gene_methylation_density_by_individual_and_season.png', bbox_inches='tight')
plt.show()
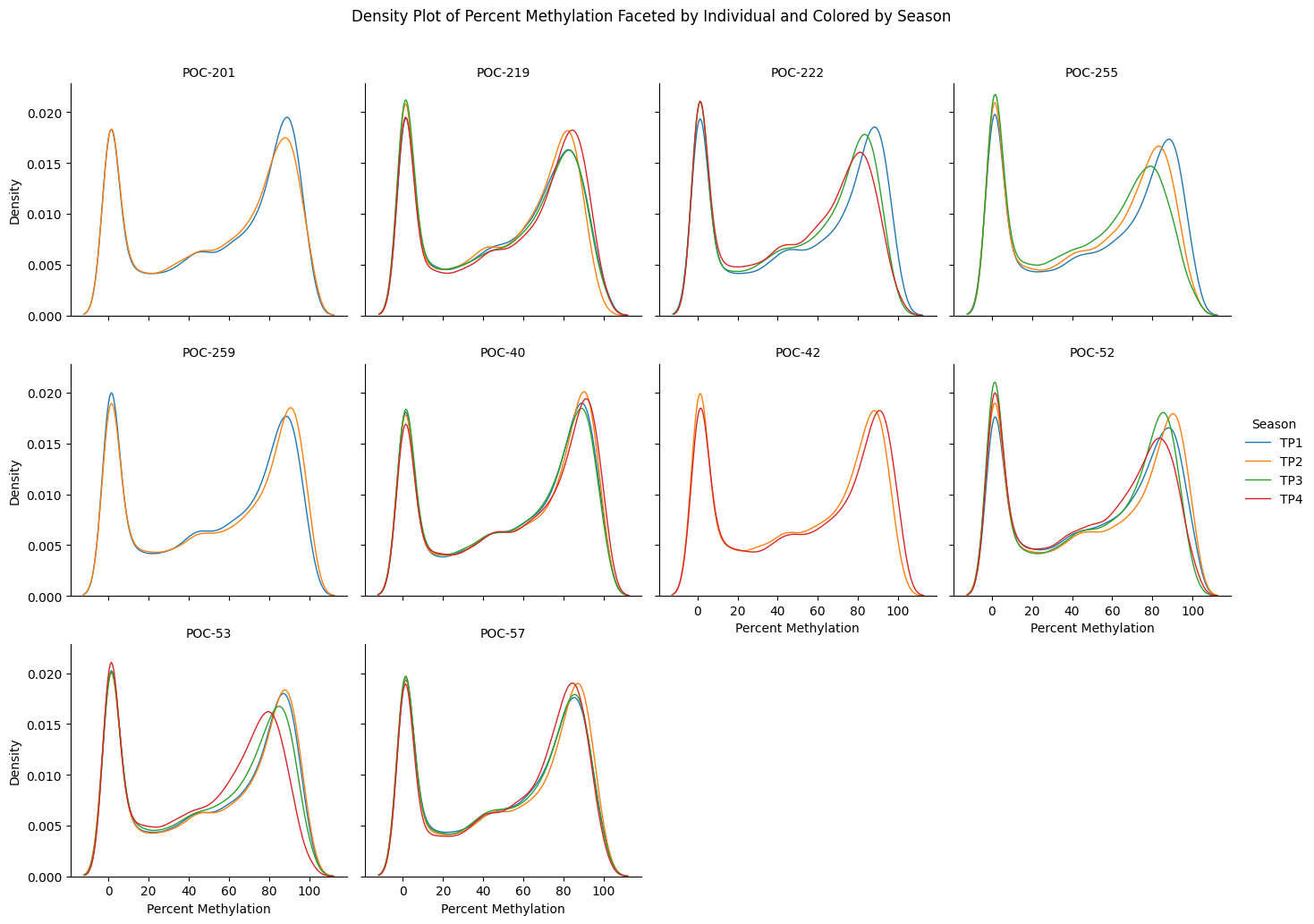
#!/usr/bin/env python3
import pandas as pd
import matplotlib.pyplot as plt
import seaborn as sns
import re
# Load methylation data
meth_df = pd.read_csv('Ptua_exon_merged_perc_meth_filt75.txt', sep='\t')
# Remove 'gene' column for melting
meth_long = meth_df.melt(id_vars=['gene'], var_name='Sample', value_name='Percent_Methylation')
# Remove NA values
meth_long = meth_long.dropna(subset=['Percent_Methylation'])
# Extract individual (remove -TP suffix) and season (TP suffix)
meth_long['Individual'] = meth_long['Sample'].apply(lambda x: re.sub(r'-TP\d+$', '', x))
meth_long['Season'] = meth_long['Sample'].apply(lambda x: re.search(r'(TP\d+)$', x).group(1) if re.search(r'(TP\d+)$', x) else 'NA')
# Facet by individual, color by season
g = sns.FacetGrid(meth_long, col='Individual', col_wrap=4, sharex=True, sharey=True, height=3.5, hue='Season', palette='tab10')
g.map(sns.kdeplot, 'Percent_Methylation', fill=False, linewidth=1)
g.add_legend(title='Season')
g.set_titles(col_template="{col_name}")
g.set_axis_labels('Percent Methylation', 'Density')
plt.subplots_adjust(top=0.9)
g.fig.suptitle('Density Plot of Percent Methylation Faceted by Individual and Colored by Season')
plt.savefig('exon_methylation_density_by_individual_and_season.png', bbox_inches='tight')
plt.show()