BSseq method clarification
- determined the following during conference call:
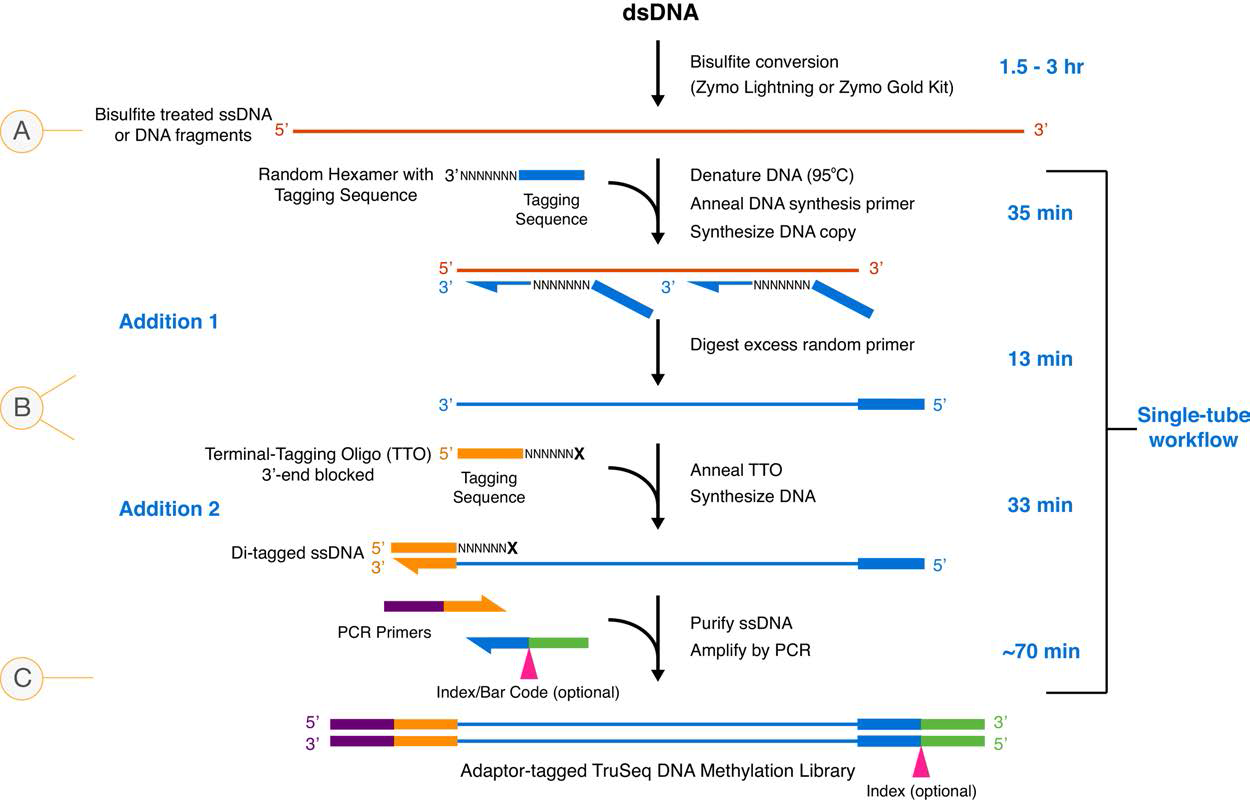
- samples were digested with MspI but not size selected prior to library prep
- digested genomic DNA was randomly primed during first round of PCR in library kit (see kit schematic)
- all read start sites should be now be random and no longer at the MspI cut site
- we DO want to deduplicate because we do not expect reads to stack up since they are randomly primed
- we should follow bismark user guide recommendations for TruSeq DNA-Methylatin Kit for trimming
- trim an extra 8bp off both ends of reads
{kind=link}
New Trimming Pilot
- Script here: 20200319_TG_EPI-Test.sh
- Slurm file here: slurm-2358892.out
- Parameter description:
-
8 bp were clipped off the beginning and end of each read using the following TG parameters:
--clip_R1 8 --clip_R2 8 --three_prime_clip_R1 8 --three_prime_clip_R2 8 -
adapters were either specified using the illumina sequences provided on page 45 of the Illumina adapter sequences document or were identified using the default settings
#specifying adapters --adapter AGATCGGAAGAGCACACGTCTGAAC --adapter2 AGATCGGAAGAGCGTCGTGTAGGGA #default settings did not include the --adapter and --adapter2 options
-
- newly trimmed reads are on Gannet here https://gannet.fish.washington.edu/metacarcinus/Pgenerosa/analyses/20200319/ and mox here:
/gscratch/scrubbed/strigg/analyses/20200319
- reads with specified adapters trimmed:
- reads with TrimGalore! defaults adapter trimmed:
- FastQC files:
- BEFORE (raw data):
- AFTER trimming with adapter specified):
- AFTER trimming with default adapters:
- Aligned reads
- Assciated github issues: - https://github.com/hputnam/Geoduck_Meth/issues/3 - https://github.com/RobertsLab/resources/issues/860
New Trimming on all data
- Pilot trimming looked good and there wasn’t a big difference between specifying the exact adapter sequence or using default adapter settings so we decided to go with default
- copied all raw data to mox: https://gannet.fish.washington.edu/metacarcinus/Pgenerosa/analyses/20200320/readme.txt
- Ran this script on mox: 20200320_TrimGpgnrMeth1.sh
- slurm file
- Trimmed reads are here: https://gannet.fish.washington.edu/metacarcinus/Pgenerosa/analyses/20200320/TG_FASTQS/
- FastQC files are here: https://gannet.fish.washington.edu/metacarcinus/Pgenerosa/analyses/20200320/TG_FASTQS/FastQC/
- MultiQC report is here: https://gannet.fish.washington.edu/metacarcinus/Pgenerosa/analyses/20200320/multiqc_report.html
- OFS was updated with newly trimmed reads https://github.com/hputnam/Geoduck_Meth/issues/4
Alignments on newly trimmed data
- Steven ran this Bismark script on mox: https://github.com/hputnam/Geoduck_Meth/blob/master/code/03-bismark.sh
- slurm file
- Bismark alignments are here: https://gannet.fish.washington.edu/seashell/bu-mox/scrubbed/032120-fds/
- Bismark summary report: https://gannet.fish.washington.edu/seashell/bu-mox/scrubbed/032120-fds/bismark_summary_report.html
- OFS was updated with new alignment files https://github.com/hputnam/Geoduck_Meth/issues/4