Generate appropriate background
- I previously used within-sample DMRs filtered for coverage in 3/4 individuals/group as the background.
- HOWEVER, these did not include all sites that had the potential to be methylated
- To create a more inclusive background, I need to look at all CG sites considered prior to determining within-sample DMRs
- Within-sample DMRs were determined by:
- having at least 3 differentially methylated sites (DMS)
- DMS were determined by:
- having 5x coverage
- if sites are within a 30bp window their counts can be combined
- passing an RMS test significance threshold of 0.01
- having 5x coverage
- DMS were determined by:
- within a max distance of 250bp
- having at least 3 differentially methylated sites (DMS)
- ATTEMPT 1: If I remove the significance threshold (and set –sig-cutoff to 1 instead of 0.01) for DMS, than I should get results from all sites considered for DMS.
- I attempted this using this mox script: 20191108_DMRfindAllEPInoSig.sh
- RESULTS: Resulting files indicate some other filtering is happening because because the number of regions is the nearly the same as when I ran DMRfind with significance level set to 0.01. I had expected a much longer list of regions.
- I attempted this using this mox script: 20191108_DMRfindAllEPInoSig.sh
- ATTEMPT 2: To address if the software only consider CG sites with a value in the methylation counts column of the allc file I created new allc files with the coverage counts column also as the methylation counts column (assuming 100% methylation at every site).
- I attempted this using this mox script: 20191108_DMRfindAllEPItotCounts.sh
- allc files with the coverage counts column also as the methylation counts here: https://gannet.fish.washington.edu/metacarcinus/Pgenerosa/analyses/20191108/tot_counts_as_mC_counts/
- RESULTS: Resulting files indicate some other filtering is happening because because the number of regions is the nearly the same as when I ran DMRfind with significance level set to 0.01. I had expected a much longer list of regions.
- I attempted this using this mox script: 20191108_DMRfindAllEPItotCounts.sh
- ATTEMPT 3: To adjust other parameters in attempt to remove all filtering, I tried the following settings in this mox script here: 20191108_DMRfindAllEPItotCountsRes1.sh on ambient samples only as a test:
- –resid-cutoff 1
- –min-tests 1
- –num-sims 1
- –sig-cutoff 1
- RESULTS: resulting files indicate certain criteria in the software are not met as output contains only a header
4th times the charm!
- ATTEMPT 4: Don’t use DMRfind, just filter allc files for 5x coverage and find all CG sites in at least 3/4 samples
- Filter allc files for sites with 5x coverages and CG context
- mox script here: 20191108_filterAllc5x.sh
- input files here: https://gannet.fish.washington.edu/metacarcinus/Pgenerosa/analyses/20190925/
- output files (and slurm file) here: https://gannet.fish.washington.edu/metacarcinus/Pgenerosa/analyses/20191108/5x_allc/
- Filter CpG sites with coverage in 3/4 individuals per experimental group, concatenate group sites for each comparison (all ambient samples, all day 10 samples, all day 135 samples, and all day 145 samples), and create bed files to intersect with GFF
- mox script here: 20191108_combine_filt_allc.sh
- I successfully implemented a bash loop within a loop to call different sets of files to filter sites for each experimental group! Found this post really helpful
- Also added ‘set -ex’ to my bash script so that the slurm file records every command as it is called. slurm file here
- Input files here: https://gannet.fish.washington.edu/metacarcinus/Pgenerosa/analyses/20191108/5x_allc/
- I unzipped the input files before running the script on mox because I kept getting zcat related errors (see slurm scripts: slurm-1459988.out and slurm-1459990.out).
- Output files here: https://gannet.fish.washington.edu/metacarcinus/Pgenerosa/analyses/20191108/combine_filt_allc/
- mox script here: 20191108_combine_filt_allc.sh
- Intersect bedfiles with GFF to get genomic features for sites from each comparison
- mox script here: 20191109_EPIbkgdFeatures.sh
- GFF file here: Panopea-generosa-vv0.74.a4-merged-2019-10-07-4-46-46.gff3
- Input files here:
- Output files here: https://gannet.fish.washington.edu/metacarcinus/Pgenerosa/analyses/20191109/
- Filter allc files for sites with 5x coverages and CG context
Plot background vs. sig. DMR features
I updated the Rmarkdown file I used previously to generate a bar plot showing the proportion of features that significant DMRs vs. background sites fall into.
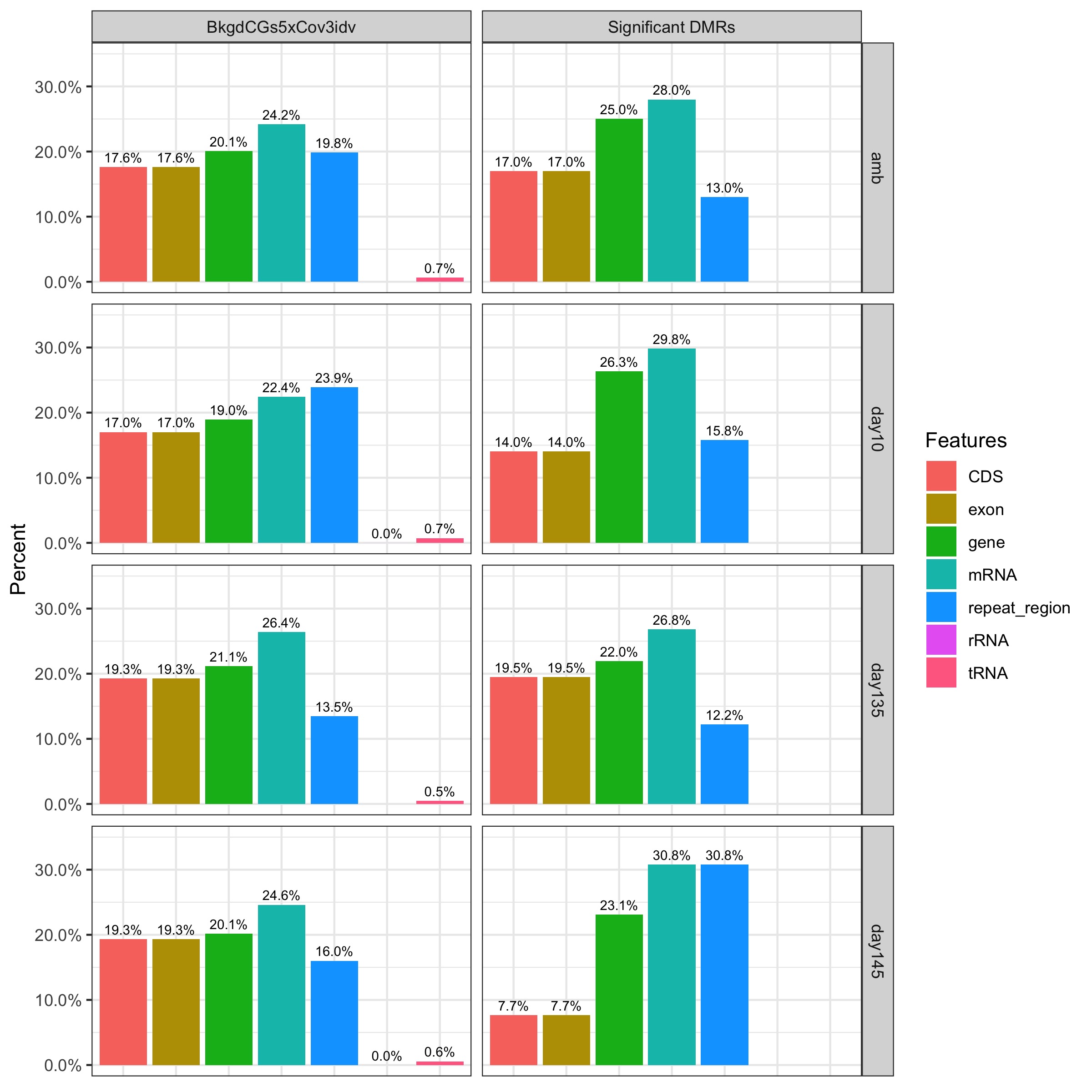
RESULTS: There are no strong differences between significant DMRs and background CpG sites in the proportion of features they overlap with, except:
-
Day145 comparison where CDS and exon features are under-represented, and repeat_regions are over-represented.
-
CDS and exon are slightly over-represented in Day135 and all ambient sample comparisons
Next steps
- Look deeper into repeat regions (there are ~6 different catagories, so can see if any are particularly different)
- For DMRs no in features, check the nearest features
- Compare genes with DMRs to the ones identified by Hollie’s method
- Continue GO analysis
- generate appropriate GO background to use for each comparison
- run TopGO
- Go through manuscript methods section
- update new stuff
- add comments to areas of concern