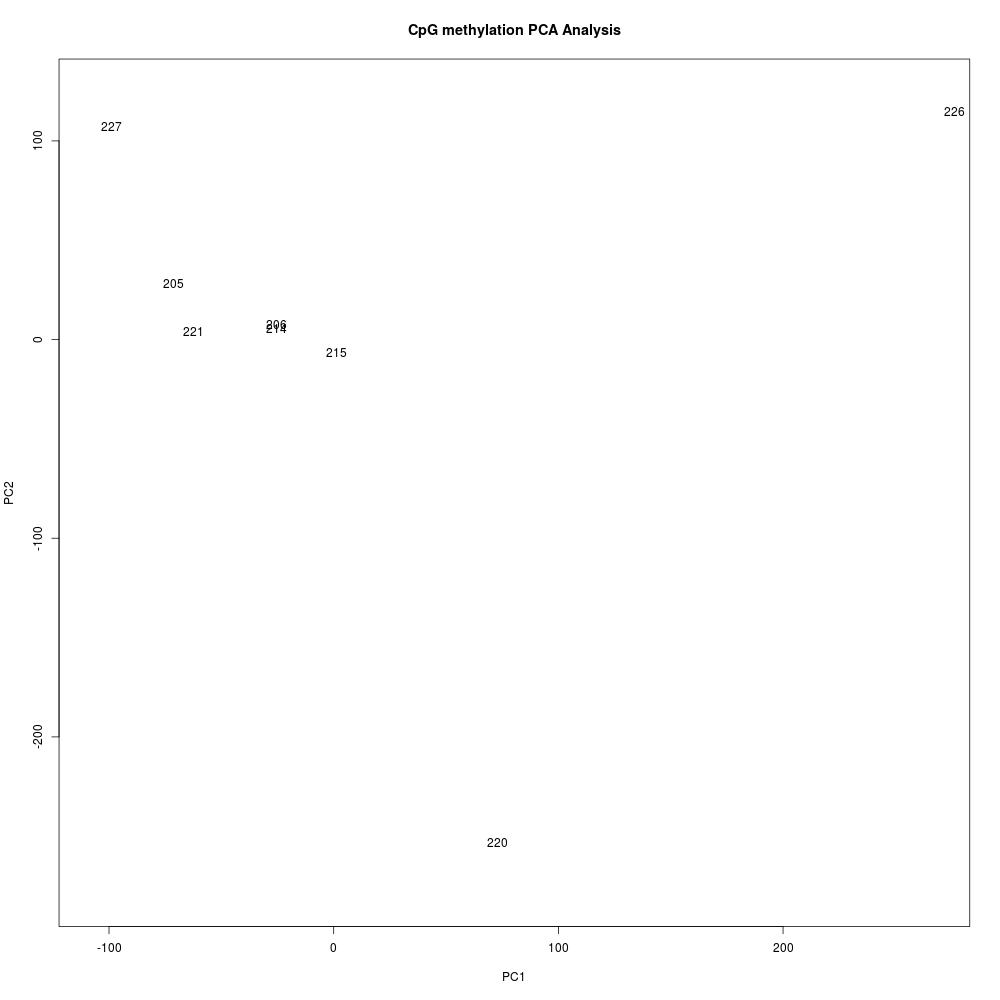
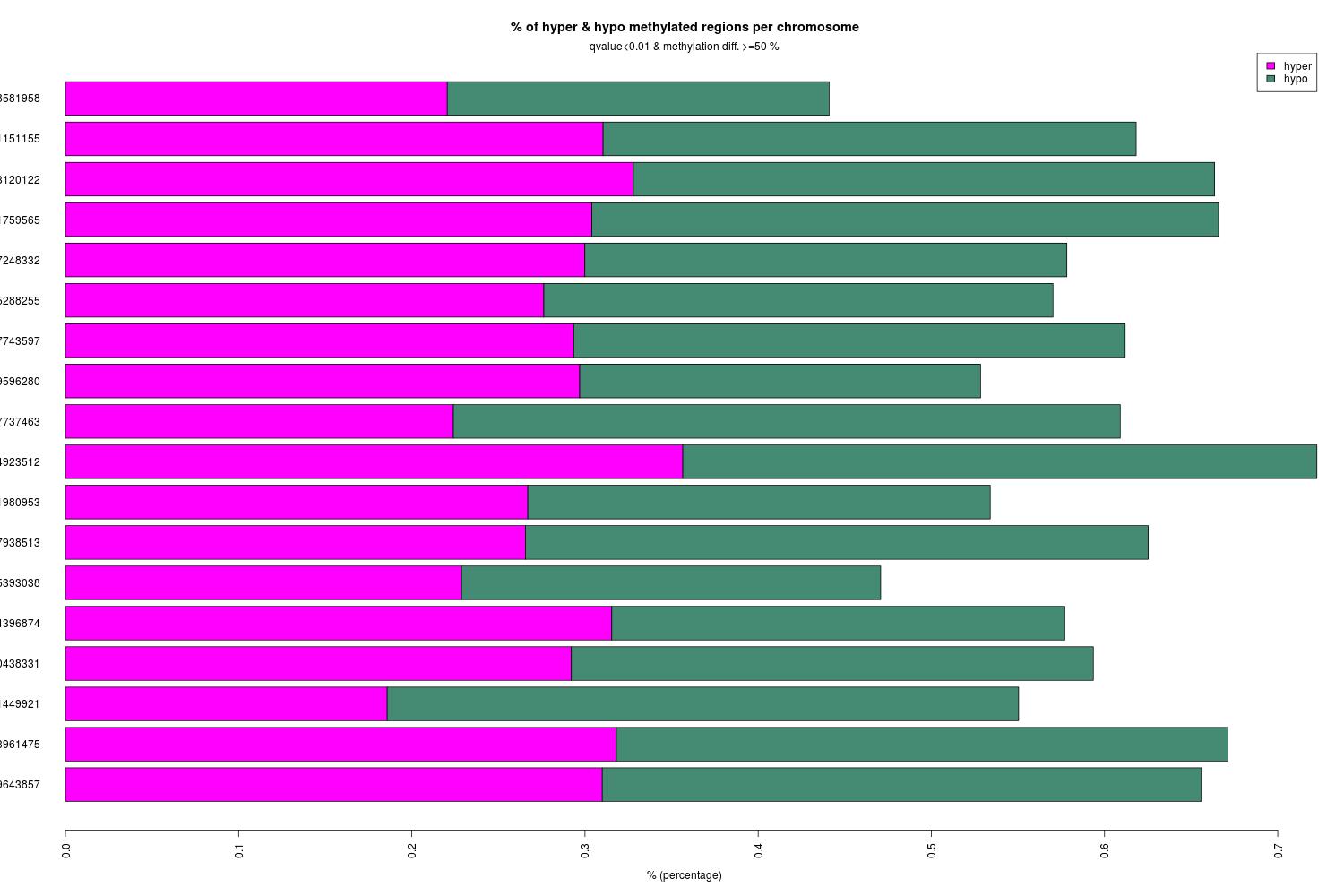
Methylkit analysis of steven’s alignments
-
copy data to emu
srlab@emu:~/GitHub/Shelly_Pgenerosa/analyses/JuviPgen_ALSL2lowd145/Get_DMRs_for_SR_v074_alignments/$ mkdir dedup_bams srlab@emu:~/GitHub/Shelly_Pgenerosa/analyses/JuviPgen_ALSL2lowd145/Get_DMRs_for_SR_v074_alignments/dedup_bams$ cd dedup_bams/ srlab@emu:~/GitHub/Shelly_Pgenerosa/analyses/JuviPgen_ALSL2lowd145/Get_DMRs_for_SR_v074_alignments/dedup_bams$ wget https://gannet.fish.washington.edu/seashell/bu-mox/scrubbed/0807-004/EPI-205_S26_L004_R1_001_val_1_bismark_bt2_pe.deduplicated.sorted.bam srlab@emu:~/GitHub/Shelly_Pgenerosa/analyses/JuviPgen_ALSL2lowd145/Get_DMRs_for_SR_v074_alignments/dedup_bams$ wget https://gannet.fish.washington.edu/seashell/bu-mox/scrubbed/0807-004/EPI-206_S27_L004_R1_001_val_1_bismark_bt2_pe.deduplicated.sorted.bam srlab@emu:~/GitHub/Shelly_Pgenerosa/analyses/JuviPgen_ALSL2lowd145/Get_DMRs_for_SR_v074_alignments/dedup_bams$ wget https://gannet.fish.washington.edu/seashell/bu-mox/scrubbed/0807-004/EPI-214_S30_L004_R1_001_val_1_bismark_bt2_pe.deduplicated.sorted.bam srlab@emu:~/GitHub/Shelly_Pgenerosa/analyses/JuviPgen_ALSL2lowd145/Get_DMRs_for_SR_v074_alignments/dedup_bams$ wget https://gannet.fish.washington.edu/seashell/bu-mox/scrubbed/0807-004/EPI-215_S31_L004_R1_001_val_1_bismark_bt2_pe.deduplicated.sorted.bam srlab@emu:~/GitHub/Shelly_Pgenerosa/analyses/JuviPgen_ALSL2lowd145/Get_DMRs_for_SR_v074_alignments/dedup_bams$ wget https://gannet.fish.washington.edu/seashell/bu-mox/scrubbed/0807-004/EPI-220_S32_L004_R1_001_val_1_bismark_bt2_pe.deduplicated.sorted.bam srlab@emu:~/GitHub/Shelly_Pgenerosa/analyses/JuviPgen_ALSL2lowd145/Get_DMRs_for_SR_v074_alignments/dedup_bams$ wget https://gannet.fish.washington.edu/seashell/bu-mox/scrubbed/0807-004/EPI-221_S33_L004_R1_001_val_1_bismark_bt2_pe.deduplicated.sorted.bam srlab@emu:~/GitHub/Shelly_Pgenerosa/analyses/JuviPgen_ALSL2lowd145/Get_DMRs_for_SR_v074_alignments/dedup_bams$ wget https://gannet.fish.washington.edu/seashell/bu-mox/scrubbed/0807-004/EPI-226_S34_L004_R1_001_val_1_bismark_bt2_pe.deduplicated.sorted.bam srlab@emu:~/GitHub/Shelly_Pgenerosa/analyses/JuviPgen_ALSL2lowd145/Get_DMRs_for_SR_v074_alignments/dedup_bams$ wget https://gannet.fish.washington.edu/seashell/bu-mox/scrubbed/0807-004/EPI-227_S35_L004_R1_001_val_1_bismark_bt2_pe.deduplicated.sorted.bam -
Create new methylkit R project and perform analysis
- R project here: Get_DMRs_for_SR_v074_alignments
- R script here: Get_DMRs_for_SR_v074_alignments.Rmd
IGV analysis
- Yaamini gave me a quick tutorial about how she used IGV to visualize differential methylation (thanks Yaamini!)
- Jupyter notebook for preparation of files to load into IGV here: 20190816_Pgnrv074_DMRs_in_IGV.ipynb
- Summary of analysis: I did a bedtools intersect between hypo- + hyper-DMRs and coverage files (filtering for positions that have 3x coverage).
- Bedgraph coverage files generated by jupyter notebook are here: 20190816_Pgnrv074_DMRs_in_IGV
- IGV session here: 20190816_Pgnrv074_DMRs.xml
- I loaded in both percent methylation and number of C’s at each position overlapping with DMRs for samples:
- 205(amb.-low)
- 206(amb.-low)
- 214(Super.low-low)
- 215(Super.low-low)
- 220(Super.low-low)
- 221(Super.low-low)
- 226(amb.-low)
- 227 (amb.-low)
- I loaded in CDS, mRNA, and gene tracks for v074 genome.
- I loaded in DMRs (as regions)
- Here’s an examples of what the diff. meth. looks like for scaffold 9 completely zoomed out:
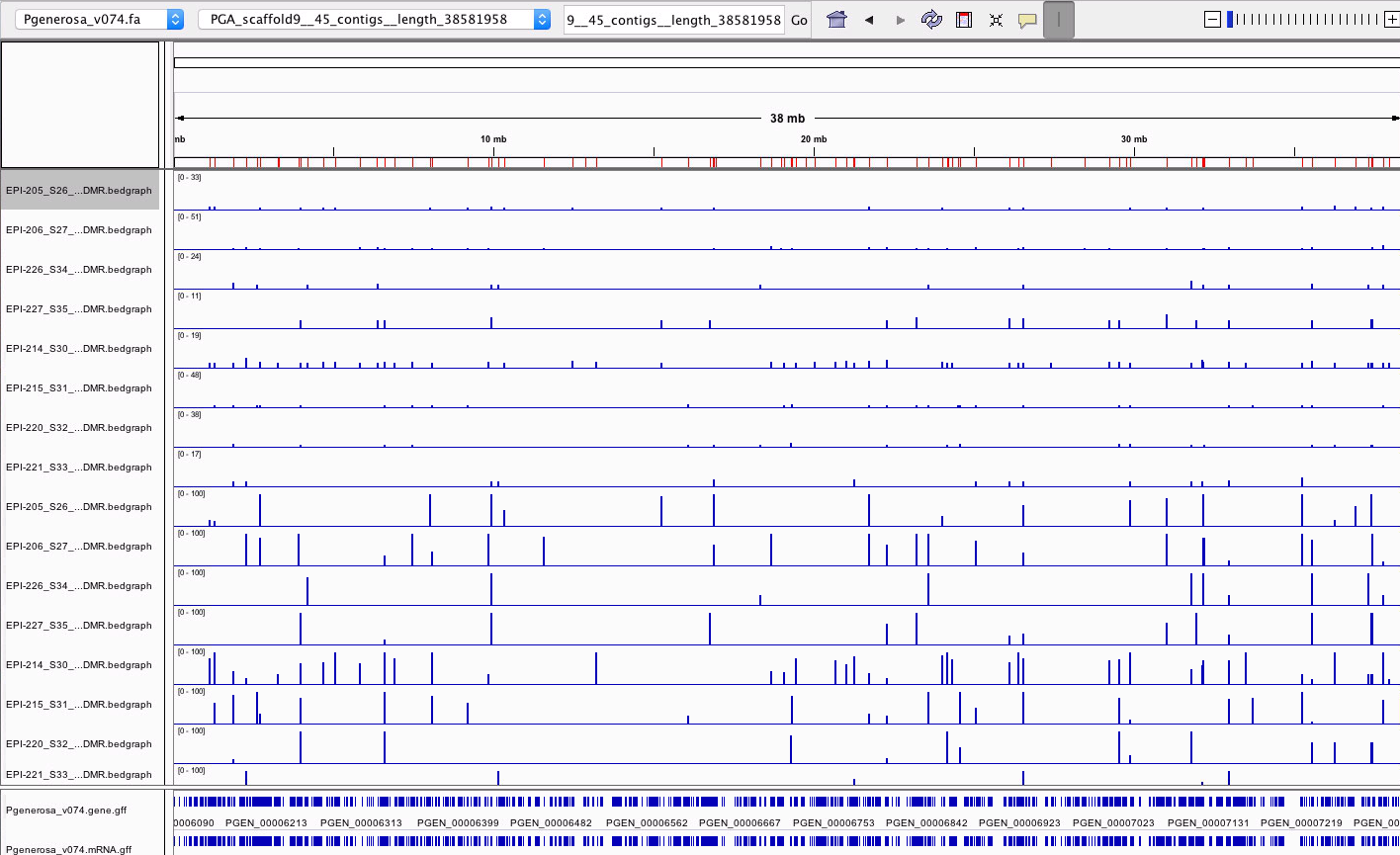
- the first 8 tracks are the number of C’s at each position. The first 4 tracks are the ambient-low group and the following 4 are the Super.low-low group.
- The bottom 8 tracks are the percent methylation at each position order the same way as the first 8 tracks.
- Here is one DMR zoomed in:

- This looks like the ambient-low group shows hypermethylation in this DMR compared to the Super.low-low group (see the first 4 of the bottom 8 tracks), HOWEVER there doesn’t seem to be any coverage of this DMR in the Super.low-low group where there is 4x coverage of it in each of the samples from the ambient-low group. So this seems like a false positive since the DMR was called because of lack of coverage. This is an issue that Yaamini encountered in here IGV diff. meth. analysis of C.virginica data
- To weed out instances like this, I need to look into:
- some kind of normalization before comparing reads?
- stricter methylkit parameters for calling DMRs
- lit on how people do DMR analysis to avoid these false positives
- I loaded in both percent methylation and number of C’s at each position overlapping with DMRs for samples:
- Summary of analysis: I did a bedtools intersect between hypo- + hyper-DMRs and coverage files (filtering for positions that have 3x coverage).