Bismark alignments
I ran this job on mox to align reads to Pgenerosa_v071.
-
Sam ran FastQC here
-
Sam mentioned Genewiz recommends “Trimming 10 bases from the beginning of both R1 and R2 following adapter trimming eliminates the majority of Adaptase tails.”
Trimming
- Instead of running trimmomatic or following Genewiz’s recommendations, I tried just doing a crude trim removing the first 6 characters (which seemed to be lower quality)from each read before running the alignments. see mox job for code
running bismark aligment and methylation extractor
- I ran bismark with score_min L,0,-1.2 and insert size minimum of 60bp (-I 60 ) on trimmed and on untrimmed reads to see if the alignments are different
- Here is the overall Bismark alignment summary report for trimmed and untrimmed reads
running cytosine coverage
- because I didn’t include the genome-wide cytosine coverage option when I initially ran bismark, I ran coverage2cytosine after with this script on mox
Calculating coverage
I adapted Sam’s new coverage script to work on my data in this jupyter notebook: 20190503_Pgnr_comparison.ipynb.
It worked! And generated this plot:
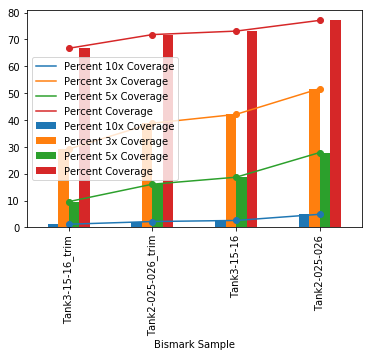
Coverage of genome-wide cytosines is surprising good. Tank 2 got about 66M reads and Tank 3 got about 50M reads. The genome is about 2.4gb. If the genome was evenly fragmented at 150bp and got even coverage, you would need 16M reads to cover the genome 1x. So it’s possible the the depth these libraries got were able to to cover most cytosines.
Next steps:
Run MethylKit
- As a first pass, run untrimmed alignments through methylkit to see what’s different
Trim properly and rerun alignments and coverage analysis
I should probably try the alignments again with the recommended trimming and see what happens
Check unmapped reads
I don’t think that Bismark outputs unmapped reads unless you specify it in your initial code: https://www.bioinformatics.babraham.ac.uk/projects/bismark/
But when I run alignments again, I’ll specify unmapped reads be output .